
If there is one thing we learned from over 150 years of studies on the evolution of sex it is that more sex is not always better. So why some bacteria exchange genetic material more frequently than others? Read the article by Dr Rafał Mostowy from the JU Małopolska Centre of Biotechnology Microbial Genomics Mostowy Lab to learn more about the reproductive mechanisms in bacteria and what they mean for evolutionary biology in general.
This article was previously published at the Mostowy Lab blog, and written on the basis of an article from Science Advances
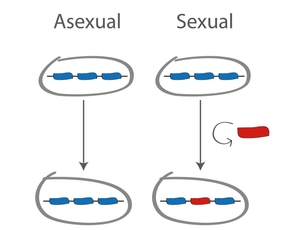
What is the best genetic strategy of making offspring? This question has been puzzling the brightest evolutionary minds for decades. One possibility is to simply make genetic copies (clones) of yourself. From an evolutionary point of view this strategy is quick, efficient and it makes sure that all of your genes are passed on to the next generation. Another possibility is to engage in some form of sex, defined as a way of shuffling or recombining your genetic material (see Figure below). Sex is a great way of acquiring new, adaptive combinations of genes, but the disadvantage is that by recombining you also risk breaking apart the adaptive combinations. So which strategy is better from an evolutionary point of view?
When asked this question, many biologists find the answer obvious: sex is better because it increases genetic diversity on which selection can act, hence accelerating adaptation of populations to changing environmental conditions. But, as neatly pointed out by Sally Otto, sex need not increase diversity and increased diversity need not be beneficial [1]. And yet almost all known organisms engage in some form of “genetic sex”. Even bdelloid rotifers, the claimed last bastion of asexuality, have been shown to sporadically exchange in some form of genetic exchange [2]. Microbes, like bacteria and viruses, evolve over time into highly mosaic genetic combination, to the extent that clonal evolution in those seems more of the exception than the rule [3-5]. So if sex and recombination are so widespread, perhaps we should not be asking 'what is the best genetic strategy of making offspring', and instead we should ask 'what rates of gene exchange are evolutionarily optimal'?
Promiscuous bacteria
Bacteria do not engage in sexual reproduction per se, but they undergo horizontal gene transfer, or HGT, which is typically defined as a combination of three distinct processes: transformation, conjugation and transduction (see image).
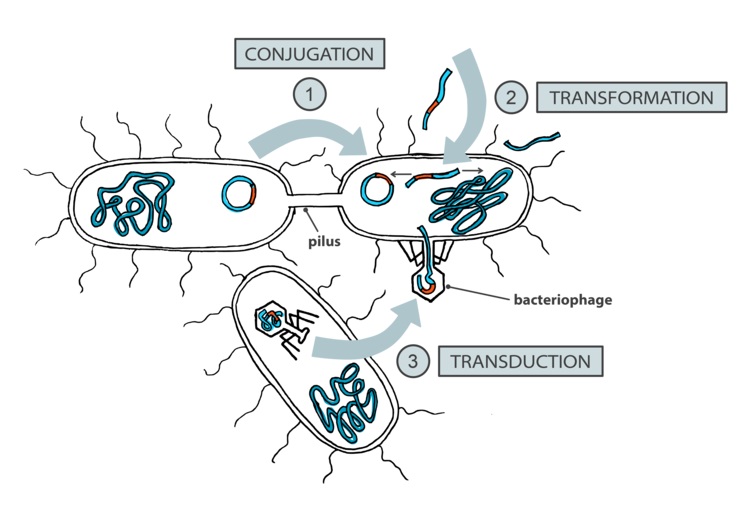
HGT is fundamentally different from eukaryotic recombination: two of the three HGT mechanisms (conjugation and transduction) are not autonomous bacterial processes, driven by the movement of selfish mobile genetic elements [6], while transformation has been argued to have evolved as a mechanism of preventing -– not enabling – the increase of genetic diversity [7]. Nevertheless, HGT has been repeatedly shown as a dominant force in bacterial evolution [8], a powerful force driving the structure of bacterial populations [9], and an important mechanism of bacterial adaptation to novel environmental niches [10]. Hence it is now commonly accepted amongst microbiologists that HGT is a genetic process tightly linked to bacterial adaptation.
The story is of course more complicated than this because bacterial populations undergo HGT at varying rates: some bacterial species, like Helicobacter pylori or Streptococcus pneumoniae, are known to recombine frequently [11]; in other bacterial species, like Mycobacterium tuberculosis, HGT is known to be extremely rare [12]. Furthermore, genomic studies have shown that within-species lineages can also vary substantially terms of the HGT rate they undergo [13]. Obviously, the HGT rate is not the only trait that can vary between bacterial lineages. So when conducting a genomic study one observes a positive correlation between HGT rate and that trait (e.g., frequency of antibiotic resistance), one is tempted to conclude that this is because HGT is adaptive and helps these lineages adapt by acquiring that trait. But if there is one thing we learned from 150 years of studies on the evolution of sex it is that more sex is not always better. So are bacterial lineages that undergo HGT at higher rates better at adapting to altered circumstances?
The pneumococcus
A few years ago I paired up with Sonja Lehtinen and Christophe Fraser (and collaborated with an incredible group of evolutionary biologists, microbiologists and epidemiologists) in an attempt to answer this question, and the results of our study have just been published in Science Advances [14]. In this paper, we have focused on S. pneumoniae (or the 'pneumococcus') which, as I mentioned above, is highly transformable. It’s been known for some time now that the pneumococcal ability to recombine helps the bacterium acquire genetic variants linked to altered response to antibiotics or vaccine [15]. Genomic studies also found a large variation in the rate of homologous recombination between global pneumococcal lineages [13]. So one question to ask is whether the more recombinogenic lineages are better at adapting. In our paper we focused on the trait of antibiotic resistance frequency, which is known to vary between different pneumococcal lineages but has also been difficult to explain in terms of standard population genetics [16]. To explain evolutionary drivers of such a variation, in our paper we proposed two hypothesis.
The first hypothesis for why some pneumococcal lineages are more antibiotic resistant than others is that they recombine more (ie., have a higher HGT rate). Previous studies had found a positive association between resistance and genetic mosaicism in different pneumococcal genes [17,18]. The proposed explanation is exactly in accordance with what I’ve been discussing here, namely that lineages that have the ability to undergo HGT more frequently are better at acquiring genes that render them antibiotic resistant, hence providing adaptation in populations where antibiotic consumption is high. We called this hypothesis 'genetics as a limiting factor'.
Now, the second hypothesis is entirely different and has nothing to do with HGT. It states that varied levels of antibiotic resistance in pneumococcal lineages reflect different selective pressures in these populations. Here’s the reasoning behind this. Pneumococci typically reside in the nasopharynx asymptomatically, a phenomenon called nasopharyngeal carriage, and some pneumococcal strains are simply better at staying there longer. Such strains with a long duration of carriage, the argument goes, are more exposed to the action of antibiotics, hence they evolve to be more antibiotic resistant. In fact, Sonja’s and Christophe’s previous work had already shown evidence of a positive relationship between the duration of carriage and antibiotic resistance [16]. We called this hypothesis 'everything is everywhere'.
Names of these two hypotheses can be better understood when you think about bacterial evolution in terms of access to genetic information and effective population sizes. If bacterial effective population size is small, then evolution becomes constrained by access to the genetic information that is available. In this case your HGT rate impacts an evolutionary outcome because a higher rate allows you to access new, adaptive gene combinations faster ('genetics as limiting factor'). However, if bacterial population sizes are very large, then evolution is not constraint by access to genetic information. In this case the HGT rate does not make such a difference because 'everything is everywhere' and 'environment can select', as stated in the famous Beijerinck’s dictum [19].
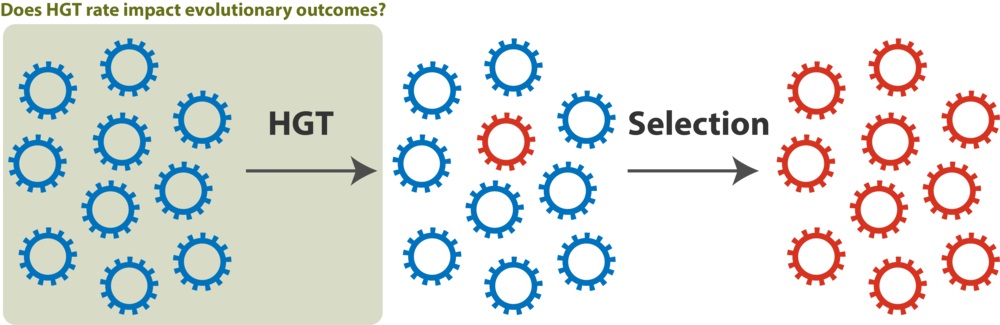
Does HGT rate influence evolutionary outcomes? If effective population sizes are small, HGT facilitates access to genetic diversity available in the population and lineages which undergo HGT more frequently will be more successful (“Genetics as limiting factor”). Conversely, if effective population sizes are large, HGT is not crucial for rapid access to genetic diversity and evolutionary success is primarily an outcome of selection (“Everything is everywhere”). In the former case what is shown in the green square is important; in the latter case it is not.
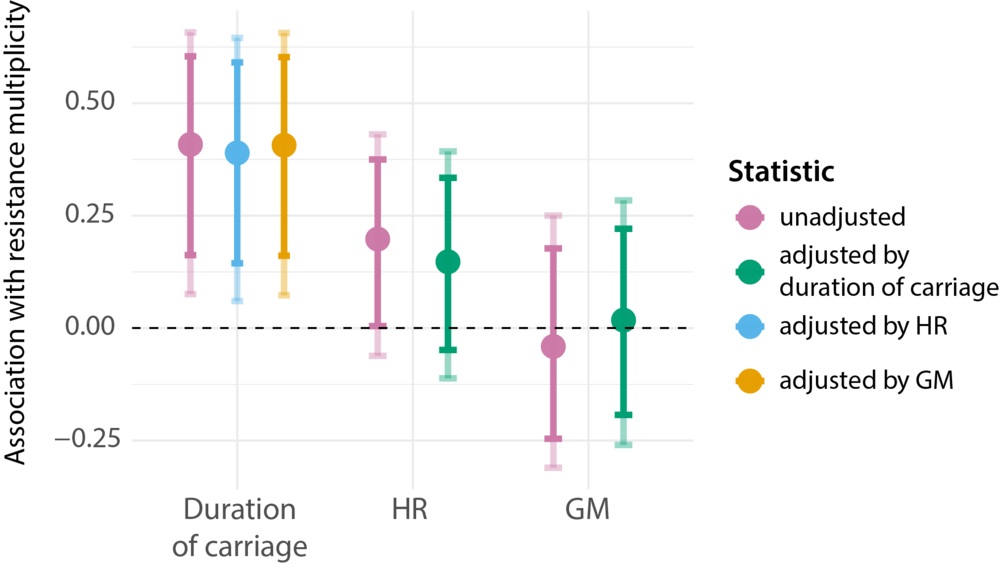
To address these two hypotheses, we analysed a collection of pneumococcal isolates from Mae La, a refugee camp on the Thai-Burmese border. Samples were isolated from mothers and babies over 3 years time, hence allowing us to identify each carriage episode and calculate its duration. The isolates were also microbiologically tested for the presence of resistance to seven classes of antibiotics. Finally, genetic sequencing of over 2,600 isolates allowed us to infer the HGT rate, measured by the horizontal transfer of alleles (homologous recombination or HR) and horizontal transfer of entire genes (gene movement or GM). All of this allowed us to make precise estimates of the antibiotic resistance frequency, duration of carriage and HGT rate for each bacterial lineage. We then applied a neat statistical approach to investigate which quantity – duration of carriage or HGT rate – better explains the variation in antibiotic resistance frequency, adjusting for one when testing for the other. You can read the details in the original publication.
Our results (see below) show a strong association between antibiotic resistance frequency and duration of carriage even when adjusting for HGT rate. We found a weak correlation between antibiotic resistance and HR rate that became insignificant when adjusting by the duration of carriage. We found no relationship between GM rate and antibiotic resistance. These findings show a strong support for the 'everything is everywhere' hypothesis, and weak support for the 'genetics as limiting factor' hypothesis.
 One really important caveat of this study is that these data were collected from a place where the pneumococcal vaccine has never been introduced. We have seen that the bacterial population there has been in a quasi-equilibrium (cf Figure S7) and found no obvious relationship between lineage frequency and HGT rate. So these results do not address a really important question about the role of HGT rate in a disruption from equilibrium, such as an introduction of the vaccine. It could be that during such an event, competition for novel ecological niches that have been freed by the vaccine provides a beneficial context for an increased HGT rate. Indeed, many organisms in nature are more likely to engage in genetic exchange in times of uncertainty [20], which makes a lot of evolutionary sense (shuffle genes when things are about to change). However, our understanding of how varying HGT rate interplays with changing environmental conditions in bacterial population is far from complete.
One really important caveat of this study is that these data were collected from a place where the pneumococcal vaccine has never been introduced. We have seen that the bacterial population there has been in a quasi-equilibrium (cf Figure S7) and found no obvious relationship between lineage frequency and HGT rate. So these results do not address a really important question about the role of HGT rate in a disruption from equilibrium, such as an introduction of the vaccine. It could be that during such an event, competition for novel ecological niches that have been freed by the vaccine provides a beneficial context for an increased HGT rate. Indeed, many organisms in nature are more likely to engage in genetic exchange in times of uncertainty [20], which makes a lot of evolutionary sense (shuffle genes when things are about to change). However, our understanding of how varying HGT rate interplays with changing environmental conditions in bacterial population is far from complete.
Conclusions
One important conclusion from all this is not the most controversial one, namely that correlation is not causation. In genomic studies where HGT rates are inferred, positive correlations between HGT rate and a given trait should not be immediately interpreted as evidence of adaptive value of HGT. We of course know that HGT is a powerful evolutionary tool, but being more prone to gene exchange itself is not always beneficial. These results also show how little we still understand about the impact of HGT rate on evolutionary dynamics of bacterial populations and the evolution of HGT rate itself. The rapid increase in size of genomic databases offers an incredible opportunity to investigate these questions. However, with millions of genomes around the corner, there is a temptation to reach for the low-hanging fruit and report on observations inferred from newly sequenced data rather than to better understand associations between previously estimated quantities. The latter is challenging as it will require novel, smarter statistical approaches (e.g., to infer causal associations or test for spurious correlations), complementary insights into the evolutionary population dynamics (e.g., from mathematical models or computer simulations) and ultimately experimental verifications of hypotheses generated using computational approaches. I think we will need all of these to provide a coherent intellectual framework to understand the role of HGT in bacterial adaptation.
References:
[1] Otto SP. The evolutionary enigma of sex. Am Nat. 2009;174 Suppl 1(S1):S1-S14. doi:10.1086/599084.
[2] Schwander T. Evolution: The End of an Ancient Asexual Scandal. Curr Biol. 2016;26(6):R233-R235. doi:10.1016/j.cub.2016.01.034.
[3] Sakoparnig T., Field C., van Nimwegen E. Whole genome phylogenies reflect long-tailed distributions of recombination rates in many bacterial species. bioRxiv 601914; doi: https://doi.org/10.1101/601914
[4] Shapiro BJ. How clonal are bacteria over time? Curr Opin Microbiol. 2016;31:116-123. doi:10.1016/j.mib.2016.03.013.
[5] Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol. 2008;25(4):762-777. doi:10.1093/molbev/msn023.
[6] Hall JPJ, Brockhurst MA, Harrison E. Sampling the mobile gene pool: innovation via horizontal gene transfer in bacteria. Philos T Roy Soc B. 2017;372(1735). doi:10.1098/rstb.2016.0424.
[7] Croucher NJ, Mostowy R, Wymant C, Turner P, Bentley SD, Fraser C. Horizontal DNA Transfer Mechanisms of Bacteria as Weapons of Intragenomic Conflict. PLoS Biol. 2016;14(3):e1002394. doi:10.1371/journal.pbio.1002394.
[8] Koonin EV. Horizontal gene transfer: essentiality and evolvability in prokaryotes, and roles in evolutionary transitions. F1000Res. 2016;5(1805):1805. doi:10.12688/f1000research.8737.1.
[9] Hanage WP. Not So Simple After All: Bacteria, Their Population Genetics, and Recombination. Cold Spring Harb Perspect Biol. April 2016. doi:10.1101/cshperspect.a018069.
[10] Sheppard SK, Guttman DS, Fitzgerald JR. Population genomics of bacterial host adaptation. Nat Rev Genet. 2018;19(9):549-565. doi:10.1038/s41576-018-0032-z.
[11] Vos M, Didelot X. A comparison of homologous recombination rates in bacteria and archaea. The ISME Journal. 2009;3(2):199-208. doi:10.1038/ismej.2008.93.
[12] Liu X, Gutacker MM, Musser JM, Fu Y-X. Evidence for recombination in Mycobacterium tuberculosis. The Journal of Bacteriology. 2006;188(23):8169-8177. doi:10.1128/JB.01062-06.
[13] Croucher NJ, Finkelstein JA, Pelton SI, et al. Population genomics of post-vaccine changes in pneumococcal epidemiology. May 2013. doi:10.1038/ng.2625.
[14] Lehtinen, Sonja, Claire Chewapreecha, John Lees, William P. Hanage, Marc Lipsitch, Nicholas J. Croucher, Stephen D. Bentley, Paul Turner, Christophe Fraser, and Rafał J. Mostowy. "Horizontal gene transfer rate is not the primary determinant of observed antibiotic resistance frequencies in Streptococcus pneumoniae." Science Advances 6, no. 21 (2020): eaaz6137.
[15] Croucher NJ, Harris SR, Fraser C, et al. Rapid pneumococcal evolution in response to clinical interventions. Science. 2011;331(6016):430-434. doi:10.1126/science.1198545.
[16] Lehtinen S, Blanquart F, Croucher NJ, Turner P, Lipsitch M, Fraser C. Evolution of antibiotic resistance is linked to any genetic mechanism affecting bacterial duration of carriage. Proceedings of the National Academy of Sciences. 2017;114(5):1075-1080. doi:10.1073/pnas.1617849114.
[17] Hanage WP, Fraser C, Tang J, Connor TR, Corander J. Hyper-recombination, diversity, and antibiotic resistance in pneumococcus. Science. 2009;324(5933):1454-1457. doi:10.1126/science.1171908.
[18] Gladstone RA, Lo SW, Lees JA, et al. International genomic definition of pneumococcal lineages, to contextualise disease, antibiotic resistance and vaccine impact. EBioMedicine. April 2019. doi:10.1016/j.ebiom.2019.04.021.
[19] O’Malley MA, ‘Everything is everywhere: but the environment selects’: Ubiquitous distribution and ecological determinism in microbial biogeography. Stud. Hist. Philos. Biol. Biomed. Sci. 39, 314–325 (2008)
[20] Ram Y, Hadany L. Condition-dependent sex: who does it, when and why? Philos T Roy Soc B. 2016;371(1706). doi:10.1098/rstb.2015.0539.
Original text: Mostowy Lab



